Phenylalanine And Tyrosine Metabolism, Urine Odors
May 22, 2023


Metabolism of Phenylaniline
Phenylalanine is converted to tyrosine (hydroxylated phenylalanine) catalyzed by phenylalanine hydroxylase.All hydroxylases are monooxygenases. Every hydroxylase will allow an oxygen molecule to react with the substrate. When an oxygen molecule reacts with Phenylaniline, one 'O' of the oxygen molecule will be incorporated into the substrate. Finally, the substrate is converted to the hydroxylated form. The other O will become water (hydrogen atoms are obtained from the co-enzyme tetrahydrobiopterin (BH2) that is converted to dihydrobiopterin)
Phenylalanine on Transamination forms phenylpyruvate. Phenylpyruvate on reduction becomes phenyl lactate.
On reduction, it is converted to phenylacetate. Phenylpyruvate (phenyl ketone) on oxidative decarboxylation forms phenylacetate. It reaches the liver and undergoes conjugation with glutamine (phenylacetylglutamine) excreted through urine.
Read this blog further to get a quick overview of this important topic for biochemistry and ace your NEET PG exam preparation.
Tyrosine Metabolism
Reaction Enzyme Tyrosine (hydroxy phenylalanine) converted to P hydroxy phenylpyruvate Tyrosine transaminase. P hydroxy phenylpyruvate to homogentisic acid. P hydroxy phenylpyruvate dioxygenase Homogentisic acid is converted to maleylacetoacetate Homogentisate oxidase Maleylacetoacetate to fumarylacetoacetate. Cis trans isomerase Fumarylacetoacetate is converted to fumarate and acetoacetate Fumarylacetoacetate hydrolase
- Amino acids, which are both ketogenic and glucogenic are phenylalanine, tyrosine, and tryptophan (aromatic amino acids)
Disorders of Tyrosine Metabolism
Type-I tyrosinemia (hepato renal syndrome)
There is Deficiency of the enzyme Fumarylacetoacetate hydrolase. Fumarylacetoacetate (precursor) accumulates which is toxic to both liver and kidney parenchymal cells (Fanconi syndrome progresses to renal failure). It results in jaundice, hepatomegaly, hypoglycemia, HCC and liver cirrhosisFanconi ,syndrome,Renal failure. Additionally, Fumarylacetoacetate is converted to succinyl acetone that inhibits the delta ALA dehydratase (heme synthetic enzyme). Elevation of succinyl acetone is considered as a pathognomonic feature. Many times, this disease is misdiagnosed as porphyria (due to neuropsychiatric manifestations).
Type-II tyrosinemia
Also known as Occulo cutaneous syndrome, Richner-Hanhart syndrome. In this there is Deficiency of enzyme: Tyrosine transaminase.Ocular changes which we are present are Painful corneal erosions and Cutaneous changes which are present are Palmar hyperkeratosis.
Type-III tyrosinemia
In this there is Deficiency of enzyme P hydroxy phenylpyruvate dioxygenase.it can be seen in patients with Recurrent seizures, intermittent ataxia. Gain of function mutation of P hydroxy phenylpyruvate dioxygenase causes hawkinsinuria (swimming pool odor of urine).
Alkaptonuria (Black urine disease)
There is Deficiency of Homogentisate oxidase. Homogentisic acid will be accumulated in the blood (undergoes oxidation to form benzoquinone acetate. It polymerizes to form melanin-like fibers (pigmentation of the skin, mucus membrane).
Osler's sign: Black or brown pigmentation of sclera between the medial and lateral rectus (sign) It accumulates in cartilage (cartilage destruction). Finally, it causes ochronosis (a feature of Alkaptonuria). Patients are normal till mid-age. Indications are multiple joint involvements, multiple intervertebral disc bulges, knee pain.Symptom include Urine turns dark on standing. Treatment is done by inhibiting the oxidation (antioxidants: Vitamin C, Nitisinone)
Nitisinone
Acts by inhibiting P hydroxy phenylpyruvate dioxygenase. Loss of gene mutations causes type-III tyrosinemia. Gain of function mutation causes hawkinsinuria. This drug was first used in the treatment of type-I tyrosinemia. Because when this enzyme is inhibited, the precursor fumarylacetoacetate is not formed. Hence, not toxic to liver and kidney parenchymal cells. It is used To treat alkaptonuria (accumulation of homogentisic acid is inhibited) and To treat hawkinsinuria. It is contraindicated in Type-II tyrosinemia
Neurotransmitters: Dopamine, Norepinephrine, Epinephrine
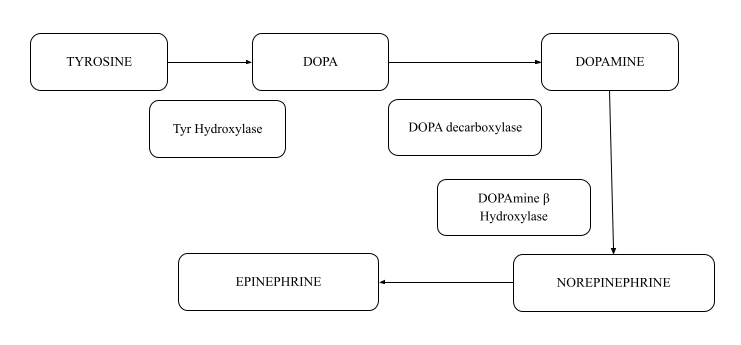
Reaction Enzyme Tyrosine to dihydroxy phenylalanine (DOPA) Tyrosine hydroxylase DOPA to Dopamine DOPA Decarboxylase (uses PLP as a coenzyme) Dopamine gives Norepinephrine Dopamine beta-hydroxylase (depends on Vitamin C) Norepinephrine forms epinephrine S adenosyl l methionine
Synthesis of Melanin Pigment
| Reaction | Enzyme |
| Tyrosine to DOPA | Tyrosine hydroxylase/ Tyrosinase |
| DOPA to Melanin | Non-enzyme-catalyzed chemical reaction |
- Tyrosinase enzyme defect causes albinism.
.jpg)
Synthesis of Thyroid Hormone
Thyroid gland is organized in the form of follicles (have a central colloid). It is surrounded by a layer of follicular cells (active protein synthesizing machinery). Protein: Thyroglobulin (synthesized)
- It is a glycoprotein containing 5000 amino acids (around 123 are tyrosine residues)
- Tyrosine residues undergo iodination to form monoiodo and diiodo tyrosine.
- It undergoes coupling to form thyronine hormones.
| During Coupling | |
| T4 | Predominant |
| T3 (the active form of thyroid hormone) | Less |
| Reversal T3 | Least |
Also Read : Electron Transport Chain
Phenylketonuria
It occurs due to Defect in Phenylalanine metabolism (major fate- Phenylalanine hydroxylase)

- Accumulation of phenylalanine.
- Diverted to minor route.
- Phenyl ketones are synthesized.
- As the phenyl ketones are accumulated, it is called phenylketonuria.
Clinical and Biochemical Features of Phenylketonuria
Characterized with
- Mental retardation (decrease in tyrosine)
- Another reason is increase in the accumulation of phenyl ketones.
- These will compete with other neutral amino acids to cross BBB.
- This results in decreased blood development.
- Phenylalanine restricted diet along with tyrosine supplements (avoid mental retardation).
- Mousy odor (accumulation of phenylacetate)
- Hypopigmentation (decreased synthesis of melanin)
Screening Tests
- Ferric chloride test: Urine (non-specific finding)-Ferric chloride forms an adsorption complex with phenyl ketones (bluish or greenish precipitate)
- Guthrie's test: Blood-Organism: Bacillus subtilis needs phenyl ketones for its growth
Take a colony plate of Bacillus subtilis.
↓
Add drop of patient’s blood
↓
After incubating, count the number of colonies.
↓
There is steady state increase of bacillus subtilis.
↓
It indicates the presence of phenyl ketones in the blood
Urine Odor and Diagnosis
Odor Disorders Mousy PKU Fruity DKA (diabetic ketoacidosis) Cabbage Tyrosinemia (type I) Boiled cabbage Hypermethioninemia Oast house (urine and sweat smell as oasthouse) or beer baby syndrome Methionine malabsorption Swimming pool Hawkinsinuria Sweaty feet Isovaleric acidemia Tom cat Urine Multiple carboxylase deficiency Fish odor Trimethyl amine urea
This is everything that you need to know about phenylalanine and tyrosine metabolism for your biochemistry preparation. For more interesting and informative blog posts like this download the PrepLadder App and keep reading our blog!

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Navigate Quickly
Metabolism of Phenylaniline
Tyrosine Metabolism
Disorders of Tyrosine Metabolism
Type-I tyrosinemia (hepato renal syndrome)
Type-II tyrosinemia
Type-III tyrosinemia
Alkaptonuria (Black urine disease)
Neurotransmitters: Dopamine, Norepinephrine, Epinephrine
Synthesis of Thyroid Hormone
Phenylketonuria
Clinical and Biochemical Features of Phenylketonuria
Urine Odor and Diagnosis
Top searching words
The most popular search terms used by aspirants
- NEET PG Biochemistry