Congenital Adrenal Hyperplasia Related Disorders
Feb 28, 2025

Basics of Adrenal Gland
There are two adrenal glands in the human body Also called suprarenal glands. Covered by a capsule Inside the capsule, they are divided into two parts:
- Derived separately from different layers.
- The two parts are the cortex and medulla.
The cortex is derived from the mesoderm. The chromaffin cells of the medulla are derived from neuroectodermal cells. The cortex is divided into Zona Glomerulosa, Zona fasciculata, and Zona Reticularis. The medulla is responsible for forming catecholamines like epinephrine and norepinephrine. The adrenal-cortical hormones are synthesized and derived from cholesterol.
Microscopic Section
Zona Glomerulosa Present just below the capsule.It is responsible for the secretion of mineralocorticoids like aldosterone. Zona fasciculata Thickest or most pre-dominant zoneIt secretes glucocorticoids like cortisol. Zona Reticularis Surrounds the medulla.It is concerned with the secretion of sex hormones.
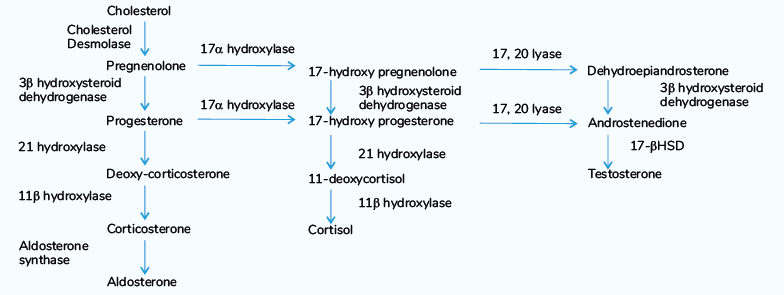
- Cholesterol is acted upon by the enzyme Cholesterol Desmolase
- Cholesterol Desmolase removes the side chain and forms Pregnenolone.
- Also known as Side-Chain Splitting enzyme
- The pregnenolone can undergo any one of the pathways to form mineralocorticoids, glucocorticoids, or sex hormones.
- Whenever there is a block or shortage of one enzyme, the cycle is disrupted.
- The intermediates present will get diverted to other pathways
- Leading to the overproduction of other hormones.
- This spectrum is called congenital adrenal hyperplasia.
Also read: Congenital and Acquired Hypothyroidism in Children
Regulation of Adrenal Cortical Hormone Synthesis
- Cortisol secretion is under the control of ACTH (corticotropin) hormone.
- Cortisol has a negative feedback inhibition on ACTH
- ACTH has positive feedback inhibition on the secretion of cortisol.
- ACTH is produced by the pituitary gland
- Controlled by CRH (corticotropin-releasing hormone) produced from hypothalamus.
- Aldosterone is controlled by:
- RAAS system (renin angiotensin aldosterone system)- most important regulator
- Serum potassium levels
- ACTH hormone (acute and mild effect)
- Sex hormone regulation is slightly complicated and not yet elucidated.
- There is a minor effect of ACTH.
Most common Adrenal cortical hormone produced in the fetus
DHEA and its derivatives are the most common hormones produced in the fetus. In the fetus, there is no significant production of aldosterone and cortisol hormones. When cortisol production increases It acts as a premature- release of surfactant from its stored site and can start the process of childbirth. So, until late gestation, the fetus has no cortisol production.
Adrenarche
The maturational process in the adrenal gland. Results in increased adrenal androgen secretion between the ages of 5 and 20 years. It begins before the earliest signs of puberty and continues throughout the years puberty is occurring. Histologically, it is associated with the appearance of Zona Reticularis.
Rate-limiting step of adrenal steroidogenesis
The importation of cholesterol across the mitochondrial outer and inner membranes. This requires several proteins, particularly the steroidogenic acute regulatory (StAR) protein. Many enzymes in adrenal cortical hormone synthesis belong to the cytochrome P450 family. The important enzymes are:
- The cholesterol desmolase enzyme is CYP11A1.
- The 21-hydroxylase enzyme is called CYP21.
- The 11-beta hydroxylase is called CYP11B1.
Enzymes 17 alpha-hydroxylase and 3 beta-hydroxysteroid dehydrogenase are not categorized as Cytochrome P450 enzymes.
Congenital Adrenal Hyperplasia
It is a group of metabolic disorders characterized by a deficiency of enzymes involved in adrenal cortical hormone synthesis. The effects are produced either due to a deficiency of one set of hormones or the accumulation of intermediates. They all show autosomal recessive inheritance. It is called hyperplasia because there is an increase in the size and thickness of the adrenal cortex. In most cases, there is a Deficiency of cortisol
↓
Loss of the inhibitory effect on ACTH
↓
Leads to a large number of ACTH hormones being produced
↓
Hyperplasia of the adrenal gland.
Large amounts of ACTH also stimulate melanocytes, and these patients are prone to hyperpigmentation. Hyperpigmentation is more common in males than in females. It is usually observed in the scrotal region.
Types of CAH (99% of cases)
- 21 hydroxylase deficiency:
- The most common type of CAH
- Seen in 90 % of the cases.
- 11 beta-hydroxylase deficiency:
- Second most common CAH
- Seen in 4-8 % of cases.
- 3-beta hydroxysteroid dehydrogenase deficiency
- 17-alpha hydroxylase deficiency
Also read: Autoimmune Polyglandular Syndromes And Ipex Syndrome
Rare Variants of CAH
Lipoid Hyperplasia: It is due to the deficiency of the StAR protein. Antler-Bixler syndrome
Clinical Presentation in CAH
All the variants of CAH are prone to Hypoglycemia due to Cortisol Deficiency. There will be either the presence of excess or deficit mineralocorticoids. Excess of mineralocorticoids produces a salt-retaining crisis. This will lead to hypertension, hypernatremia, and hypokalemia. A deficiency of mineralocorticoids causes a salt-wasting crisis. This will lead to shock, hyponatremia, and hyperkalemia. The genitalia of patients can be normal or ambiguous.
- Males will have Micro-penis, Bifid Scrotum and Undescended Testis. So it will look female.
- Females will have Clitoromegaly and Labial Fusion.
- Previously known as Pseudohermaphrodite
.png)
21-Hydroxylase Deficiency
Epidemiology and its Types
- It comprises more than 90 % of all CAH cases.
- Types of 21 hydroxylase deficiency
- Classic diseases:
- Salt-wasting form: It is the most severe form
- Simple virilizing form: Virilization of females is usually seen.
- Non-classic disease:
- It is asymptomatic
- Many cases do not come to attention till adulthood.
- Classic diseases:
- The incidence of the classic disease is 1 in 15000 to 1 in 20000 live births.
- The incidence of the non-classic disease is 1 in 500 to 1 in 1000 live births.
Also read: Maternal Diabetes And Neonatal outcomes
Genetics
- 21-Hydroxylase enzyme is also called the CYP21 gene and is present on chromosome 6p21.3. It is specifically present between HLA-B and HLA-D R-alleles.
- CYP21P is present between genes coding for C4A and C4B complement proteins, CYP21P is considered a pseudogene as it is 90 % similar to the CYP21 gene. CYP21P does not code for any protein as it has more than 300 mutations.
- CYP21 is present between C4B and TNX-B genes. True gene
- More than 90 % of the mutant alleles causing 21-hydroxylase deficiency result from recombination between CYP21P and CYP21 gene. Out of these mutations, 20 % are deletions due to unequal meiotic crossovers. The remaining are non-reciprocal transfers of deleterious mutations, also called gene conversion.
- The deleterious mutations of CYP21P have different effects on enzymatic activity when transferred to CYP21. Complete absence of the activity of this gene → severe deficiency of the gene, Partial absence of activity of the gene → relatively mild deficiency
Genotype-Phenotype correlation
Mutation group A B C Enzyme Activity No enzyme activity 1-2% normal activity 20-50% normal activity Incidence 1/20000 1/50000 1/500 Severity Salt-wasting Simple virilizing Non-classic type Age at Diagnosis Infancy Infancy in females and childhood in males. Childhood to adulthood. It may be asymptomatic, also. Aldosterone synthesis Low Normal Normal Virilization Severe. Moderate to mild None to mild
Also read: Important MCQ's Pediatric Pulmonology & Infections
Pathogenesis
- In case of deficiency of the 21-hydroxylase enzyme
- Progesterone will not get converted to Deoxy cortisone
- 17-dehydroxy progesterone will not get converted to 11- deoxycortisol
- Cortisol deficiency will lead to hypoglycemia.
- Aldosterone deficiency will lead to a salt-wasting crisis.
- Since these two cycles are stopped, their precursors will produce excess testosterone hormone.
- Will cause in-utero virilization in females → Ambiguous genitalia
- In males, there will be normal Genitalia but later in life there will be Precocious Puberty.
Clinical Features
- There will be hypoglycemia in patients.
- There will be a salt-wasting crisis leading to shock, hyperkalemia, and hyponatremia in the blood.
- In utero, virilization in Females (Ambiguous Genitalia) and normal genitalia in males.
- Postnatally, in females:
- The internal genitalia is female-type
- But because of excess androgen, this can lead to menstrual irregularities like amenorrhea
- Hirsutism, acne, obesity, and male-like voices
- Postnatally, in males
- Precocious puberty and premature closure of the growth plate leading to short stature
- Acne, obesity, and hoarseness-like voices
- Some degree of aggression can also be seen in patients of both genders.
Also read: Important MCQ's in Pediatric Endocrinology & Rheumatology
Problems of Females with Severe 21 Hydroxylase Deficiency
- Females with severe deficiency are found to have severe gender identification problems.
- Gender Dysphoria is not common unless there is severe virilization.
- Most of them are found to be heterosexual.
- They have a higher tendency to develop homosexuality.
- They have a higher tendency to develop aggressive behavior,
- They are found to have a relative lack of maternal instincts.
Adrenomedullary Dysfunction
Some patients with the severe 21-hydroxylase deficiency will have a decreased exercise response to epinephrine and norepinephrine. Decreased responses of tissues to maintain glucose levels in response to epinephrine. They have an increased risk of developing bradycardia in relative epinephrine deficiency.
Screening Test and Diagnosis
- The screening test checks the levels of 17-hydroxyprogesterone levels.
- If the levels are ≥ 1000 nanograms/ml → 21-hydroxylase deficiency.
- It can also be done as a dried blood spot test. Confirmation of this diagnosis is done by the ACTH stimulation test - Gold's standard test. The baseline of 17-hydroxyprogesterone is checked. Cosyntropin injection is given to the patient, and the levels of 17-hydroxyprogesterone are re-checked after an hour. The results are plotted on a normogram. If the values are significantly high and there is a standard deviation above the mean, it is a positive ACTH stimulation test.
Treatment
Glucocorticoid replacement is considered the mainstay of therapy. The drug of choice is hydrocortisone. It should be given lifelong. The dosage is 15-20mg/m /day in three divided doses. In case of stress, illness, or surgery, the dose doubles or triples. Monitoring of hydrocortisone therapy Serial height and weight measurements are done frequently. If it is found that despite therapy, the child's height is above +2 standard deviations, then undertreatment is suspected in the child. If the height is dipping below -2 S.D. or there is an increase in weight, then overtreatment is suspected. The serum 17-OH progesterone levels are checked. Serum Androstenedione levels are checked. For mineralocorticoid replacement, the drug of choice is Fludrocortisone. The dosage is 0.1-0.2 mg/day in infancy and 0.05 mg/day in childhood. Supplemental sodium is needed in these patients as they show a salt-wasting crisis.
11 Beta-Hydroxylase Deficiency
It comprises 4-8% of all CAH cases. Nelson: 5% average. It is the most common CAH associated with hypertension. The most common hormonal cause of hypertension in infancy. Most forms are severe and classic. The mild forms are rarely seen.
Treatment
Lower doses of Hydrocortisone are the therapy of choice for these individuals. If BP does not improve, then calcium channel blockers are started. For Ambiguous genitalia – treatment similar to that of 21-hydroxylase deficiency.
17 Alpha-Hydroxylase Deficiency
It comprises < 1% of cases of CAH. It is most common in China and Brazil. The gene is present on chromosome 10q24.3. There is no testosterone or cortisol formation. Females will have normal genitalia, Males will have ambiguous genitalia. Cortisol deficiency will cause hypoglycemia. There will be raised levels of deoxy-corticosterone and aldosterone, and this will cause a salt-retaining crisis. Since high levels of deoxy-corticosterone inhibit renin, then Patients will have low aldosterone levels, and deoxy-corticosterone levels are high → Leading to hypertension. Since corticosterone has cortisol-like action, Hypoglycemia in these children is least common among all CAH. In isolated 17, 20 lyase deficiency will have only genitalia changes.
Treatment
Hydrocortisone is the drug of choice for treatment. (Lower doses). Hormone therapy with or without surgery is given to males only if they come to attention in early life. Because of the possibility of malignant transformation of abdominal testes, Genetic males with severe 17-hydroxylase deficiency being reared as a female requires gonadectomy at or before adolescence.
3 β-Hydroxysteroid Dehydrogenase Deficiency
It comprises < 2% of all cases of CAH. 3 β-HSD is required for the conversion of Δ-5 steroids (pregnenolone, 17 hydroxy-pregnenolone, DHEA) to Δ-4 steroids. (progesterone, 17-dehydroxyprogesterone and androstenedione). The gene is present on chromosome 1p13.1. There will be a salt-wasting crisis and hypoglycemia. DHEA is a weak androgen, and it is present in high levels thus, Males and females both will develop ambiguous genitalia, It will also produce virilization in females.
Treatment
Hydrocortisone is the drug of choice, and it is given lifelong. Fludrocortisone and supplemented sodium are also given. Hormone therapy with or without surgery in case of ambiguous genitalia (patient-specific)
Lipoid Adrenal Hypoplasia
It is very rare and is most reported in Japan. Etiology: In most cases, it is the mutations of the StAR protein. StAR protein that promotes the movement of cholesterol from the outer to the inner mitochondrial membrane. If there is a mutation, cholesterol will not get converted into its various forms, This will lead to cholesterol accumulating in the cells → death of Adrenocortical cells. In some cases, there may be a mutation in the CYP11A1 gene, which encodes the cholesterol side chain cleavage enzyme, cholesterol desmolase.
Treatment
Hydrocortisone is the drug of choice. Fludrocortisone is also administered. All will be reared as females, Gonadectomy needs to be performed in males. Female hormonal support is required post-puberty.
Deficiency of P450 Oxidoreductase (POR)
Also called Antley-Bixler syndrome. The POR gene is present on chromosome 7q11.3. It is needed for the activity of enzymes CYP17 (17 alpha-hydroxylase) and CYP21 (21-hydroxylase) as well as other P450 microsomal enzymes. A single recurrent mutation A287P (alanine -287 to proline) is found on approximately 40 % of the alleles.
Important MCQ’s
Q. A 25-day-old male infant is brought to the emergency with complaints of vomiting, seizures, and poor feeding. Examination reveals a micropenis and undescended testis with a bifid scrotum. Investigations were ordered and revealed random blood glucose of 28mg/dl, sodium of 126meq/l, and serum potassium of 6.1meq/l. BUN and serum creatine were in the normal range. USG revealed a cerebriform pattern in both adrenal glands bilaterally. One previous sibling had a history of developing similar problems and dying in infancy. What is the likely diagnosis?
A. Familial glucocorticoid deficiency
B. Wolman syndrome
C. Anti-21 hydroxylase antibodies
D. Deficiency of 3-beta hydroxysteroid dehydrogenase
Explanation:
The above-given symptoms fulfill the diagnosis of 3-beta hydroxysteroid dehydrogenase deficiency. Anti-21 hydroxylase antibodies can cause autoimmune Addison's disease in some children There will be no ambiguous genitalia. Wolman syndrome is a disorder whose hallmark is adrenal calcification. Familial glucocorticoid deficiency patients will have isolated low cortisol, and ultrasound will show adrenal gland atrophy.
Q. A 3-week-old male child has features of hypoglycemia, shock, hyponatremia and hyperkalemia, and normal genitalia. Which is the most likely diagnosis?
A. 21- hydroxylase deficiency
B. 11 beta-hydroxylase deficiency
C. 17 alpha-hydroxylase deficiency
D. 3 beta- HSD deficiency
Q. A 3-week-old female child has features of hypoglycemia, salt-wasting, and ambiguous genitalia. Her 17-OH progesterone levels are inappropriately low. Which among the following is likely to be elevated?
A. Testosterone
B. Androstenedione
C. DHEA
Hope you found this blog helpful for your NEET SS Pediatric Endocrinology preparation. For more informative and interesting posts like these, keep reading PrepLadder’s blogs.

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Navigate Quickly
Basics of Adrenal Gland
Microscopic Section
Regulation of Adrenal Cortical Hormone Synthesis
Most common Adrenal cortical hormone produced in the fetus
Adrenarche
Rate-limiting step of adrenal steroidogenesis
Congenital Adrenal Hyperplasia
Types of CAH (99% of cases)
Rare Variants of CAH
Clinical Presentation in CAH
21-Hydroxylase Deficiency
Epidemiology and its Types
Genetics
Genotype-Phenotype correlation
Pathogenesis
Clinical Features
Problems of Females with Severe 21 Hydroxylase Deficiency
Adrenomedullary Dysfunction
Screening Test and Diagnosis
Treatment
11 Beta-Hydroxylase Deficiency
Treatment
17 Alpha-Hydroxylase Deficiency
Treatment
3 β-Hydroxysteroid Dehydrogenase Deficiency
Treatment
Lipoid Adrenal Hypoplasia
Treatment