Adrenal Gland: Anatomy, Function, and Disorders
Feb 9, 2024

Anatomy
Blood Supply- Adrenal glands are paired glands situated at the retroperitoneum around the superior and the medial aspect of the kidney. They are situated are the level of the 11th rib. Weight of each adrenal gland is 4 grams. It is supplied by branches of 3 important vessels: Superior adrenal artery- it is a branch of inferior phrenic artery. Middle adrenal artery- it is a branch of abdominal aorta. Inferior artery-it is the branch of the renal artery. Venous drainage occurs through a large single vein on both sides. Venous drainage on the right side occurs through the large right adrenal vein which drains into the inferior vena cava. Venous drainage on the left side occurs through the large left adrenal vein which drains into the left renal vein.
Physiology
The outer part of the adrenal gland is the adrenal cortex. It comprises 90% of the adrenal gland. It is made up of: Zona glomerulosa- produces aldosterone. Zona Fasciculata- produces cortisol . Zona reticularis- produces androgen. The inner part of the adrenal gland is called the adrenal medulla. It contains chromaffin cells; these stain yellow with chromium salt. It synthesises catecholamines adrenaline, nor-adrenaline, dopamine . The adrenal medulla contains an enzyme called phenylethanolamine-N-methyltransferase which catalyses the conversion of adrenaline to nor-adrenaline.
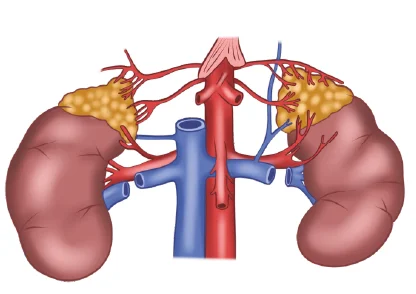
Evaluation Of The Adrenal Gland
Imaging Techniques
- Findings in a CT- Density- 10 HU, Size > 4cm. It has homogenous, irregular, or ill-defined borders. When given a contrast- Washout is <50%, relative washout <40%, absolute washout <60%, after giving contrast, there is delayed attenuation of >35 HU. Other features that can be seen are necrosis, internal hemorrhage, calcification. Findings in MRI-T2 weighted imaging- Adenoma- it has a low signal intensity as compared to the liver. Carcinoma- it has a moderate signal intensity as compared to the liver. Pheochromocytoma (PCC)- it has extremely high signal intensity; mass: liver is 3, appears like a ‘light bulb pattern’. MRI chemical shift- It is not consistent with the findings of lipid-containing adenoma- There is no loss of signal on out-of-phase imaging. Other imaging- Radionucleotide imaging with NP-59, Uptake of NP-59 100% predictive of benign adenomatous lesion. Lack of uptake of NP-59 rules out adenomatous lesion
- FDG PET- Highly sensitive and specific for diagnosing malignancy. However, it cannot differentiate between an adenocarcinoma and metastasis.
- FNAC- A CT-guided FNAC should be done. However, this is not routinely indicated. FNAC cannot cytologically diagnose a malignancy. When should it be considered? If there is a history of malignancy and one is suspecting adrenal metastasis. If it is a solitary adrenal metastasis. Before considering CT-guided FNAC, PCC should be ruled out because it can precipitate a hypertensive crisis.
Treatment And Management
If it is a functional tumor, surgery or adrenalectomy is considered. The Standard of care is laparoscopic adrenalectomy . If it is a non-functional tumor- the tumor size is <4cm with no suspicious findings in the imaging study.
Surgery is not recommended. Patient is under surveillance or follow-up. Repeat imaging is done after 3-6 months. Annual MRI is done for 1 to 2 years. If the lesion size does not increase, the patient can be followed-up. If there is >20% increase in the tumor size, with at least >5 cm increase in the largest dimension-surgery and adrenalectomy should be considered.
During follow-up if the non-functional tumor becomes hormonally active or there is autonomous hormonal production from the tumor, surgery and adrenalectomy should be considered. If the tumor is >3 cm, there is a chance that it can become hyper-functional; if these become active during follow-up- surgery and adrenalectomy should be considered.
If the tumor is >6 cm, there is a >25% risk of malignancy - surgery and ADRENALECTOMY should be considered. If the tumor is 4-6 cm with suspicious findings on imaging-surgery and adrenalectomy should be considered. If there is an adrenal mass with suspicion of carcinoma with a diameter <6cm without e/o local invasion/suspected lymph nodes, laparoscopic adrenalectomy should be done. If there is suspicion of carcinoma with e/o local invasion/metastatic lymph nodes of any size, open adrenalectomy should be done. If the >6 cm with suspicious of malignancy but no local invasion-Surgical intervention should be individualised; preferably an open surgery is considered.
Also Read: Head Injury : Introduction, Fractures of Brain, Management of Fractures

Adrenocortical Carcinoma
It is a rare but aggressive malignancy. Commonly seen in the age group of 40-50 years. It is equally seen in males and females. At presentation, 50% of the ACC are presented as functional tumours . The most common presentation is Cushing syndrome and virilization. Around 30% to 40% of the patients may present with abdominal mass or pain, back pain. Usually the tumor is large in size i.e. 9 to 10 cm and presented at an advanced stage. Metastasis can be in the lymph nodes, liver, and lungs.
Evaluation
Rule out the functional status- Clinical examination should be done thoroughly to rule out symptoms and signs of glucocorticoid excess. Other biochemical tests include- dexamethasone separation test, serum adrenal androgen (DHEAS, androstenedione, testosterone, 17 hydroxy progesterone) plus serum oestradiol in men plus post-menopausal women. Aldosterone and plasma renin ratio and potassium levels assessment in hypertensive patients, steroid precursors in 24 urine and plasma metanephrines
- Radiographic evaluation - CT- Heterogenous mass with irregular or indistinct borders, central necrosis with invasion of adjacent structures . FDG-PET- Can rule out occult metastasis. Maximum SUV>40 or >1.5 times the liver is diagnostic of malignant tumors. Only definitive criteria for distinguishing between benign and malignant is by checking for the presence of distant metastasis or local invasions. When one is unable to differentiate between benign and malignant tumors, modified Wiess histopathological system is used: >6 mitoses /50 HPF- given score 2, <= 25% clear tumor cells in the cytoplasm- given score 2, Abnormal mitosis- given score 1, Capsular invasion- given score 1, Necrosis- given a score 1. A total score of 3 or >3 is suggestive of a malignant tumor.
Treatment And Management
Treatment includes surgery i.e. R0 resection. One may have to consider resection of primary tumor En bloc resection. If there is a local invasion with the adjacent organs, radical lymphadenectomy must be done . At presentation, there may be 3 scenarios- Indeterminant /probably malignant tumour <6 cm-laparoscopic adrenalectomy should be done . If local invasion on imaging or suspicion of it at laparoscopy- open adrenalectomy Y should be done. Indeterminant or probably malignant tumor> 6cm- preferably open adrenalectomy is considered Indeterminant or probably malignant tumor with synchronous metastatic disease. If it is limited metastatic disease-resection of the primary tumor and the metastasis .If it is a widespread metastasis-systemic therapy should be considered
- Systemic therapies used in adrenocortical carcinoma- Mitatone- it is a derivative of DDT and is toxic to adrenocortical carcinoma. It kills tumors with an increased chance of recurrence, non-resectable and metastatic
- Which tumors are at high risk of recurrence? which are >5 cm in size , with an increase in Ki67, rupture at the time of surgery, presence of thrombosis . In all the above cases, metatone should be started and continued for 5 years. It improves the disease-free and overall survival. Patients not responsive to the above remedy can be given palliative EDP chemotherapy i.e. Etoposide-doxorubicin-cisplatin CT
Adrenal Metastasis
Primaries that can cause metastasis to the adrenal gland are lung cancer, breast cancer, colorectal cancer, RCC, melanomas. When adrenal metastasis occurs, 50% of them are bilateral. This can cause adrenal insufficiency; 30% of the patients can present with this. Adrenal insufficiency can be a differential diagnosis of adrenal incidentaloma.
Treatment And Management
Management depends on whether they are Isolated adrenal metastasis or widespread metastasis. Isolated adrenal metastasis occurs from renal cell carcinoma, lung cancer, and colorectal cancer. With widespread metastasis, surgery is not considered, instead palliative or systemic therapy is considered.
Evaluation
CT thorax plus and plus PET CET- to rule out extra-adrenal disease. Evaluate for adrenal insufficiency-assess the levels of morning cortisol and ACTH. Screening of catecholamine plus cortisol excess- To rule out a coincidental hormonal functional tumor
Surgery
- If adrenal metastasis is diagnosed with 1/L renal cell carcinoma- adrenalectomy can be done at the time of nephrectomy. In case of other primary tumors- a patient is observed and scan is repeated after 3 to 6 months; after 3 to 6 months of the tumor is solitary, isolated, and stable, surgery can be considered . If adrenal metastasis > 6 months after initial treatment i.e. they are metachronous, PET CT is done to see if there is solitary, isolated metastasis or a widespread disease; If isolated, surgery can be considered
- What type of surgery should be considered? If the adrenal metastasis is < 4 cm- laparoscopic adrenalectomy should be considered. If the adrenal metastasis is bigger-open adrenalectomy should be considered
Also Read: Congenital Anomalies of the Kidneys and Urinary Tract
Principles Of Adrenal Surgery
- Laparoscopic surgery
- Indications of laparoscopic adrenalectomy: Presence of functional tumors , aldosterone secreting tumor, cortisol secreting tumor, bilateral hyperplasia, symptomatic adrenal cyst or adrenal myelolipoma, adrenal incidentaloma, small ACC without local invasions. Contraindications of laparoscopic adrenalectomy - absolute contraindication- Local recurrence of previously resected adrenal mass. Invasive ACC with e/o invasion of neighbouring organs/renal vessels/ IVC. Severe cardiopulmonary disease. Relative contraindication- Large tumor (> 6 CM), presence of abdominal adhesions or surgeries, recurrent pyelonephritis, pregnancy, virilizing tumors (70% to 80% of these are ACC)
- Routine pre-operative antibiotics are used only in patients with an increased risk of infections e.g. those with Cushing’s syndrome. These patients should receive prophylactic antibiotics before the laparoscopic adrenalectomy
- Laparoscopic approaches- Transabdominal- Anterior transabdominal approach- No repositioning is required if a bilateral technique is used. Lateral transabdominal approach- Most commonly used approach by surgeons. Useful in obese patients, those with large tumors. Posterior retroperitoneal- Preferred when a patient has a history of previous abdominal surgery and when abdominal adhesions are suspected.
Open surgery
- Anterior approach- bilateral adrenalectomy can be done by a single incision . Posterior- Preferred when a patient has a history of previous abdominal surgery and when abdominal adhesions are suspected . Lateral- Useful in obese patients, those with large tumors . Thoracoabdominal- when there are big tumors and en bloc resection has to be considered
Adrenal Incidentalomas
They are adrenal masses that are clinically inapparent and incidentally detected on imaging. They include both benign and malignant tumors of the cortex and medulla. They are caused by: Non-functioning adenomas-60% cases, Pheochromocytoma - 10% of cases, Myelolipoma-9% cases , Cortisol producing adenoma-5% cases , Adrenocortical Ca- 5% cases, Metastases-2% cases. In patients with a history of extra-adrenal malignancy who suffer from adrenal incidentaloma, the most common cause is metastasis . 50% of the adrenal metastasis is bilateral. Most common malignancies that cause metastasize to the adrenal gland are Lung > Breast > Kidney. Concerns in patients with adrenal incidentaloma. If the tumor is a functional or a non-functional tumor. If the tumor is benign or malignant or If there is a history of malignancy.
Evaluation
- Clinical assessment: Look for signs and symptoms related to hypertension, glucocorticoid excess, sex steroids excess. Investigations- Low dose (1mg) overnight Dexamethasone suppression test- this is to rule out subclinical diseases such as Cushing’s syndrome. Plasma free metanephrines/24-hour urinary metanephrines + catecholamines-this is to rule out pheochromocytomas. Plasma aldosterone concentration, plasma renin activity, and serum electrolytes to be assessed in patients with hypertension to rule out aldosterone.
- If subclinical cushing’s syndrome is suspected, the following tests should be considered-Dexamethasone suppression test, 24-hour urinary cortisol levels assessment, plasma cortisol levels assessment. Findings on CT- Benign lesion has a homogenous appearance. It is well-defined, well-encapsulated, smooth, regular borders. It has a high lipid content. It is hypoattenuating < 10 HU. There is a rapid washout of contrast material because there is decreased vascularity. When should there be a suspicion of malignancy based on CT findings? If size > 4 um, hyperattenuating >18 HU, in homogenous, irregular, or ill-defined borders. Contrast-enhanced washout CT, Relative <40%, Absolute < 60%. Necrosis, internal hemorrhage, calcifications.
- Findings in MRI: T2 weighted imaging, Adenoma- it has a low signal intensity as compared to the liver. Carcinoma- it has a moderate signal intensity as compared to the liver. Pheochromocytoma (PCC)- it has extremely high signal intensity; mass: liver is 3, appears like a ‘light bulb pattern. Radionuclide imaging with NP59- not considered nowadays- If the lesion uptakes NP59, it is adenomatous lesion. If there is no uptake of NP59, it is a non-adenomatous lesion
- FDG PET: They are very specific and very sensitive to differentiate between benign and malignant lesions. It cannot differentiate between metastasis and adrenocortical malignancy
- CT guided FNAC: It is not routinely indicated in patients with adrenal. This is because primary adrenal carcinoma cannot be diagnosed based on cytology. It is indicated if patient has extra-adrenal malignancy and metastasis is suspected. Before CT-FNAC, PCC should be ruled out or else, it can precipitate a hypertensive crisis
Treatment And Management
- If the tumor is functional, adrenalectomy is considered. If the tumor is functional, it is found the tumor is benign or malignant and addresses the issue accordingly.
Also Read: RENAL STONES - Etiology, Investigation and Management
Pheochromocytoma And Parganglionoma
- Pheochromocytoma(PCC): They arise from the neural ectodermal tissue of the adrenal medulla. The tumor produces catecholamines
- Paraganglionomas(PGL): They arise from the extra-adrenal sympathetic and parasympathetic ganglia. Both Pheochromocytoma and Paraganglionomas together called PPGL
- Parasympathetic PGL: They arise from parasympathetic ganglia . 95% of them do not produce catecholamines. They are non-secreting tumors. They are also called Nonchromaffin paragangliomas. They most commonly arise in the head and neck; they are thus also called head and neck paragangliomas. The most common locations are the carotid body, vagal and jugulotympanic
- Sympathetic PGLs: They produce catecholamines. They are commonly seen in the abdomen and the pelvic place along the paravertebral and prevertebral sympathetic chain .The most common site is the organ of Zuckerkandl
- Previously Pheochromocytoma was called a 10% tumor. This is because, 10% of the PCC are bilateral, 10% are malignant, 10% are extra-adrenal, 10% are seen in children, 10% are familial
Genetics Of Pheochromocytoma
- Pheochromocytoma -75% are sporadic; 25% are familial. Paraganglionomas - Sporadic paraganglionomas are less common than sporadic pheochromocytoma. They are more associated with the hereditary endocrine system. Overall 70% of paraganglionomas are sporadic. Familial paraganglionomas occur at younger ages; they are commonly bilateral, multifocal, and malignant. Syndromes associated with familial paraganglionomas are MEN 2(2A+2B). Pheochromocytoma occurs in families with MEN2A and MEN2B in 50% of pts; the mutations seen are most commonly in codons 634 and 918. Pheochromocytoma associated with MEN2 syndrome are not malignant; only 2 to 3% are associated with MEN 2 are malignant. Other syndromes associated with familial PPGL include NF1-Neurofibromatosis, Sturge Weber syndrome, VHL- It is associated with chromosome 3P; 14% of paraganglionomas are affected with pheochromocytoma. Other manifestations of VHL: renal cyst, pancreatic cyst, RCC, hemangioblastomas, retinal angiomas. Hereditary PPGL syndrome/Familial paraganglionoma syndrome
Also Read: Prune Belly Syndrome (Eagle Belly Syndrome)
Hereditary Pheochromocytoma And Paraganglionomas Syndrome
It occurs due to mutation in the succinate dehydrogenase (SDH) gene family which mainly includes the SDH B, C, and D subunits . Mutations are most common in SDH D > SDH B. Mutations have also been described in SDHA, SDHAF2, SDH5. SDHA, B, C- It is an autosomal dominant condition. SDHD, AF2- will cause mutation in the only progeny of affected fathers; these result in hormonally inactive or non-secreting paragangliomas which are more commonly seen in the head and neck region and are associated with multiple tumors. SDH-B mutation variants- lead to secreting paragangliomas; 50% of PCC occurring from SDHB mutation are malignant . Mutation in SDHD result in hormonally inactive or non-secreting paragangliomas which are more commonly seen in the head and neck region and are associated with multiple tumors. Affected individuals should undergo regular surveillance with annual blood tests and 3 yearly MRIs of the neck and the abdomen. Other mutations associated with hereditary PPGL syndrome include- Myc associated protein X(MAX). Transmembrane protein 127 (TEM127). MAX mutations cause tumors only if inherited from the father. Germline mutations in B and D subunits of SDH can be seen in 10% of patients with sporadic PCC
Pheochromocytomas
- It is present mainly in the 4th and the 5th decade of life. The hereditary ones present 15 years before the presentation of sporadic ones. They are equally seen in males and females. They are functional tumors that secret catecholamines. Adrenal PCC mainly produces noradrenaline more than adrenaline. PGLs produce more adrenaline compared to noradrenaline. Malignant PCC is associated with increased production of dopamine. secretory PGL mainly produce noradrenaline because they lack Phenylethanolamine N-methyltransferase (PNMT) which helps in the conversion of noradrenaline to adrenaline. PCC can have a varied clinical presentation and because of this, it is also called a great masquerade. There can be a classical triad of PCC, this includes headache (most common symptom of PCC), palpitation, diaphoresis or profuse sweating; this is seen only in a selected subset of patients. Other symptoms include: anxiety and panic attacks, flushing, tremulousness, pallor, weakness, weight loss, chest pain, shortness of breath
- All the above symptoms are episodic because it corresponds to the episodic hormonal production by the tumor cells; they occur in paroxysms that last for less than an hour. Most common sign of pheochromocytoma is hypertension which is seen in 90% of patients. This is episodic and can also be sustained. On examination, patients may demonstrate tachycardia, when a patient has hypertension along with the classical triad, there is a high chance that the patient has pheochromocytoma. The signs and symptoms can be triggered by certain factors- stress, surgery- induction agents of the general anaesthesia , contrast medium, exercise , certain drugs such as tricyclic anti-depressants, Opiates, and metoclopramide
Pathology Of Pheochromocytoma
- It appears like a yellow-tan well-defined lesion- it can compress the adrenal gland. After incubation with potassium dichromate, the lesion becomes dark brown. Highly vascularized with areas of hemorrhage, necrosis, and cystic areas. Histologically- Nests of Polygonal/spindle-shaped neuroendocrine chief cells with peripheral glial sustentacular cells; this pattern is called the zell ballen pattern. Nuclei- Salt and pepper chromatin. Immunohistochemistry-it is positive for chromogranin and synaptophysin in the chief cells. in the sustentacular cells, it is positive for S-100 antigen.
Diagnosis Of Pheochromocytoma
Plasma tests: The plasma metanephrines test has a high sensitivity (>99%), meaning it is very good at identifying people who have the disease. If this test is negative, it essentially rules out the presence of a pheochromocytoma. However, the specificity of this test is somewhat lower (85-89%), meaning it may sometimes indicate pheochromocytoma in people who don't have the condition (a false-positive result). Therefore, if the test is positive, it typically needs to be confirmed with additional testing. A 24-hour urinary test for total metanephrines and catecholamines is often used to confirm the diagnosis. This test measures the number of metanephrines and catecholamines excreted in the urine over 24 hours.If this test also returns a positive result, it's a strong indication of pheochromocytoma.
- 24-hour urinary test: Total Metanephrines: The urine is collected over 24 hours and tested for total metanephrines (normetanephrine and metanephrine). This test is highly specific (93-95%), so a positive result strongly indicates the presence of a pheochromocytoma.
- Catecholamines: The 24-hour urine test for catecholamines (norepinephrine, epinephrine, and dopamine) is also a helpful diagnostic tool. Increased levels of catecholamines in the urine over 24 hours can be indicative of a pheochromocytoma.
- Fractionated Catecholamines and Metanephrines: The 24-hour urine can also be analyzed for fractionated catecholamines and metanephrines. This can provide more specific information about which hormones are being overproduced and can help differentiate between pheochromocytomas and other conditions.
- Clonidine suppression test: This is a confirmatory test that is not frequently used due to the availability of high-quality biochemical testing. However, it may be used in some specific circumstances. A baseline blood sample is taken to measure the patient’s plasma catecholamine levels. The patient is then given a dose of oral clonidine (typically 0.3 mg). Three hours after the clonidine is administered, another blood sample is taken to measure the post-clonidine plasma catecholamine levels. The result is considered positive (indicating the possible presence of a pheochromocytoma) if the plasma catecholamine levels do not fall below a certain threshold, often set at 500 pg/mL.
Localisation
- MRI: An MRI can provide detailed images of soft tissues such as the adrenal glands, where pheochromocytomas are usually located. This can help to identify the location and size of the tumor and potentially provide information regarding its characteristics. MRI is an excellent imaging modality for localizing pheochromocytoma and paragangliomas, with a sensitivity of up to 95%. The "light bulb" sign, referring to the high signal intensity seen on T2-weighted images, is a characteristic feature of pheochromocytoma due to its high-water content. Gadolinium-enhanced MRI can further improve the visibility of these tumors, particularly in detecting extra-adrenal paragangliomas, which can occur anywhere along the sympathetic and parasympathetic chains. MRI is the preferred imaging modality in certain patient populations, especially in those with contraindications to CT contrast, and as you correctly stated, in pregnant women. The ionizing radiation from CT scans can potentially harm a developing fetus, making MRI the safer option for imaging during pregnancy.
- CT scan: A CT scan is a common first-line imaging test in many centers because it is widely available, quick, and effective in identifying adrenal masses. It is reported to have a sensitivity of 85-95% and a specificity of 70-100% in detecting pheochromocytomas. A CT scan can help determine the size of the tumor and may also suggest its malignancy based on characteristics like irregular borders, invasion of surrounding structures, or presence of distant metastases. For detecting extra-adrenal pheochromocytomas, also known as paragangliomas, there are additional nuclear medicine imaging studies that can be used. These tests utilize special isotopes that bind to particular types of cells, including pheochromocytoma and paraganglioma cells.
- MIBG Scintigraphy: I-131 or I-123 labelled metaiodobenzylguanidine (MIBG) scans are often used. MIBG is a substance that is absorbed by certain types of nervous tissue, including pheochromocytomas and paragangliomas. The radioactive I-131 or I-123 isotope is detected by a special camera, which is used to visualize the location of the tumor. This scan is highly specific for pheochromocytoma but has a sensitivity of around 77-90%.
- Octreoscan (111In-labeled Octreotide Scintigraphy): Octreotide is a synthetic version of somatostatin, a hormone that naturally occurs in the body. Octreotide preferentially binds to somatostatin receptors, which are often overexpressed on the surface of neuroendocrine tumors, including paragangliomas. When labeled with Indium-111, it allows for imaging of these tumors. The sensitivity of octreotide scanning is around 50-70%.
- Positron Emission Tomography (PET) or PET/Computed Tomography (PET/CT): scanning with novel radiotracers are increasingly being used in the diagnostic evaluation of pheochromocytoma and paraganglioma, especially for lesions that are MIBG negative or in the detection of metastatic disease. 18F-DOPA PET/CT (Fluoro-L-dihydroxyphenylalanine): This is a type of PET scan that uses a radioactive form of the amino acid DOPA. 18F-Fluorodopamine PET/CT: This is another type of PET scan that uses a radioactive form of the molecule dopamine, which is taken up by pheochromocytomas and paragangliomas. These PET scans are often used when initial imaging studies, like CT/MRI and MIBG scans, are negative but clinical suspicion remains high. The most promising novel radiotracer for these types of tumors is Gallium-68 (68Ga) labeled with peptides that bind to somatostatin receptors (DOTA-coupled peptides), which are often overexpressed in these tumors. These include:
- 68Ga-DOTATATE PET/CT: The 68Ga-DOTATATE scan has shown particularly high sensitivity and specificity for detecting pheochromocytoma and paraganglioma, even higher than traditional MIBG and FDG-PET scans.
- 68Ga-DOTATOC and 68Ga-DOTANOC PET/CT: These are similar to the 68Ga-DOTATATE scan and can also be used for imaging these tumors.
- Preoperative preparation is critical in the management of patients with pheochromocytoma. The goal is to minimize the risk of hypertensive crisis or severe blood pressure fluctuations during and after surgery, which could result from the manipulation of the tumor and the consequent release of catecholamines. Additionally, a high-salt diet and fluid intake are encouraged to increase blood volume, helping to prevent hypotension following the removal of the tumor.
- Intraoperatively, careful monitoring and management of blood pressure and heart rate are necessary. There could be a significant release of catecholamines during induction of anesthesia or manipulation of the tumor, leading to a hypertensive crisis. After the tumor is removed, there can be a rapid drop in catecholamines leading to hypotension. This can be exacerbated by hypovolemia, decreased peripheral arterial resistance, and increased venous capacitance, potentially leading to cardiovascular collapse if not managed appropriately.
Preoperative Care
- Alpha-adrenergic blockade is the cornerstone of preoperative management in patients with pheochromocytoma. Here are the key points: Alpha-adrenergic blockade: Phenoxybenzamine, a non-selective and non-competitive alpha-receptor antagonist, is commonly used. By irreversibly binding to alpha-receptors, it can help to control blood pressure and prevent a hypertensive crisis during surgery. Dosing: The starting dose is usually around 10 mg twice daily, which can be increased every 2 to 3 days until the maximum tolerated dose or blood pressure and symptoms are controlled, typically not exceeding 40 mg three times daily. Duration: Treatment with phenoxybenzamine is usually started at least 2 weeks before surgery. This allows for the normalization of blood pressure and heart rate, and adequate reversal of alpha-adrenergic receptor downregulation, which restores the sensitivity of the vasculature to vasoactive agents. Side effects: The most common side effects of phenoxybenzamine include postural hypotension (a drop in blood pressure upon standing), nasal congestion, reflex tachycardia, and fatigue.
- Beta-blockers: These are used in patients with persistent tachycardia or arrhythmia after adequate alpha-blockade has been achieved. They are never initiated before alpha-blockers because unopposed alpha-stimulation can lead to a hypertensive crisis. These medications are usually introduced 2-3 days before surgery, once blood pressure is under control.
- Calcium Channel Blockers: These are added in patients who have inadequate blood pressure control even after titration of alpha-blockers. They are an alternative to beta-blockers for controlling tachycardia. The most commonly used calcium channel blockers include Nicardipine.
- Metyrosine (Demser): This is a medication that inhibits tyrosine hydroxylase, the enzyme involved in catecholamine synthesis. It is used in patients who have poor tolerance to alpha and beta-blockers or have inadequate control of blood pressure even after titration of these medications. Metyrosine can be a valuable adjunct in controlling symptoms and blood pressure in these patients, particularly in those with malignant or bilateral tumors, or in those awaiting liver transplantation for metastatic disease.
Preoperative intravascular volume expansion with isotonic fluids is not typically required when aggressive alpha blockade has been effectively implemented.
Treatment
Treatment of choice is surgery: Surgery is the preferred approach for addressing adrenal gland conditions. Surgery of choice is adrenalectomy: Adrenalectomy refers to the surgical removal of the adrenal gland. It can be performed using laparoscopy (minimally invasive) or through an open procedure.
- Laparoscopy: Laparoscopic adrenalectomy is the standard approach for surgical removal of the adrenal gland when feasible. It involves making small incisions and using a camera and specialized instruments for the procedure. Laparoscopic adrenalectomy is contraindicated: There are situations where laparoscopic adrenalectomy is not advisable. One such situation is when imaging suggests the presence of malignancy with local invasion. Large tumors may require open surgery: For relatively large tumors, specifically, those exceeding 10cm in size, the open surgical approach may be preferred over laparoscopy. Hemodynamic instability during surgery: During the surgical procedure, there may be instances where there is a sudden fall in blood pressure when the adrenal vein is clipped. To address this, the medical team should administer fluids and vasopressors to restore blood pressure. Paragangliomas: Paragangliomas (PGLs) can sometimes be more complex than pheochromocytomas due to their extra-adrenal location and potential for local invasion. Therefore, an open surgical approach may be preferred in some cases of paragangliomas.
- Postoperative Management: Following surgery, it's important to monitor patients for potential complications including hypoglycemia (low blood sugar) and hypertension. In some cases, the body takes time to adjust to the sudden drop in catecholamines after the tumor has been removed, and this could potentially lead to a period of hypotension (low blood pressure) instead of hypertension. Surveillance: Following successful removal of the tumor, patients are typically monitored with annual biochemical testing for at least five years to check for any signs of recurrence, metachronous (occurring at different times) tumors, or metastatic disease.
- Familial Pheochromocytoma: In familial cases of pheochromocytoma where there's a risk of bilateral disease, bilateral cortical-sparing adrenalectomy may be considered. In this procedure, the surgeon aims to preserve around 1/3 of the adrenal cortex, if possible. This is to avoid the need for lifelong corticosteroid replacement therapy, which would be necessary if the entire adrenal glands were removed.
Pheochromocytoma
The anesthetic management of patients undergoing surgery for pheochromocytoma is a crucial aspect of their care, given the significant risks of intraoperative hypertensive crisis and arrhythmias. Inhaled agents: Inhalation agents such as isoflurane and enflurane can be used for anesthesia as they cause minimal cardiac depression. Avoid certain agents: Certain anesthetic agents including fentanyl, ketamine, and morphine should be avoided because they can stimulate the release of catecholamines, potentially provoking a hypertensive crisis. Management of arrhythmias: If intraoperative arrhythmias occur, they can be managed with short-acting beta-blockers like esmolol. This should be done under careful monitoring because beta-blockers can potentially worsen hypertension if the alpha blockade is not adequate. Blood pressure control: Intraoperative hypertension can be managed with vasodilators such as nitroprusside. Phentolamine, an alpha-adrenergic blocker, can also be used.
Malignant Pheochromocytoma
- Most pheochromocytomas are benign, about 5-10% can be malignant. Unfortunately, distinguishing benign from malignant pheochromocytoma based solely on the tumor’s appearance at the time of surgery can be challenging, as the definitive diagnosis of malignancy is made by the presence of metastases in non-chromaffin sites (sites that are normally devoid of chromaffin cells). Here are some factors that might be suggestive of malignancy:
- Increased PASS score: The Pheochromocytoma of the Adrenal gland Scaled Score (PASS) is a tool that pathologists use to predict the potential for malignancy based on the tumor’s appearance under the microscope. A higher score is associated with an increased risk of malignancy.
- Increased Ki67: Ki67 is a protein in cells that increases when they are preparing to divide. A higher percentage of cells staining positive for Ki67 may suggest a more aggressive tumor.
- Presence of capsular invasion: This refers to the tumor growing beyond the capsule that surrounds the adrenal gland, which may suggest a higher risk of malignancy.
- Vascular invasion: The presence of tumor cells within the blood vessels is often considered suggestive of malignancy.- Malignant pheochromocytomas often produce excessive amounts of dopamine. Common sites of metastases for malignant pheochromocytomas include lymph nodes, bones, liver, and lungs.
Treatment
- The treatment of choice for malignant pheochromocytoma, when feasible, is surgical resection. The goal is to remove as much of the tumor as possible. In cases of metastatic disease, this may involve multiple surgeries or a combination of surgical and other treatment modalities. Unfortunately, malignant pheochromocytoma is relatively resistant to traditional chemotherapy and radiation therapy. However, several therapeutic approaches have been used with varying degrees of success:
- Alpha-adrenergic blockers: As with benign pheochromocytoma, these medications (such as phenoxybenzamine) are used to block the effects of excessive catecholamines produced by the tumor.
- 131I-MIBG: This is a form of targeted radionuclide therapy that uses a radioactive form of iodine that's taken up by pheochromocytoma cells. This can help to reduce the size of the tumors and alleviate symptoms.
- DOTATOC labeled with Yttrium or Lutetium: This is another form of targeted radionuclide therapy. The DOTATOC molecule binds to somatostatin receptors that are overexpressed in pheochromocytoma cells, and the attached Yttrium or Lutetium isotope delivers targeted radiation to the tumor cells.
- Chemotherapy: Although traditional chemotherapy regimens have limited effectiveness, some chemotherapy drugs can be used. For instance, Averbuch's protocol involves the use of dacarbazine, vincristine, and cyclophosphamide.
Pheochromocytoma In Pregnancy
Pheochromocytoma is a rare but potentially life-threatening condition in pregnancy, for both the mother and the fetus. The presentation can indeed be "silent" or subtle, with symptoms such as hypertension, headaches, or palpitations, which can be mistakenly attributed to normal pregnancy changes. The estimated fetal mortality rate in untreated pheochromocytoma in pregnancy is about 12%. Diagnosis and management depend on the trimester of pregnancy during which the pheochromocytoma is detected.
- Diagnosis in the 1st and 2nd Trimesters: If pheochromocytoma is diagnosed in the first or second trimester of pregnancy, medical management with alpha-blockers (and possibly beta-blockers) is initiated to control symptoms and blood pressure. This is followed by surgical removal of the tumor ideally in the second trimester, which is generally considered the safest time for surgery in pregnancy.
- Diagnosis in the 3rd Trimester: If pheochromocytoma is discovered in the third trimester of pregnancy, the management strategy depends on the gestational age and maternal-fetal status. If the near term, it may be feasible to medically manage the patient until safe delivery can be performed, followed by surgical resection of the tumor postpartum. However, if the diagnosis is made earlier in the third trimester or if there is a crisis, it may be necessary to perform a cesarean section followed immediately by an adrenalectomy.
If you are preparing for NEET-SS 2024 and ahead, check out SS ELITE Plan (Version 3.0) and what makes it the perfect study resource for your super speciality preparation.

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Navigate Quickly
Anatomy
Physiology
Evaluation Of The Adrenal Gland
Imaging Techniques
Treatment And Management
Adrenocortical Carcinoma
Evaluation
Treatment And Management
Adrenal Metastasis
Treatment And Management
Evaluation
Surgery
Principles Of Adrenal Surgery
Open surgery
Adrenal Incidentalomas
Evaluation
Treatment And Management
Pheochromocytoma And Parganglionoma
Genetics Of Pheochromocytoma
Hereditary Pheochromocytoma And Paraganglionomas Syndrome
Pheochromocytomas
Pathology Of Pheochromocytoma
Diagnosis Of Pheochromocytoma
Localisation
Preoperative Care
Treatment
Pheochromocytoma
Malignant Pheochromocytoma
Treatment
Pheochromocytoma In Pregnancy
Top searching words
The most popular search terms used by aspirants
- NEET SS Surgery Breast and Endocrine
- NEET SS Surgery Preparation Plan