Pigmentary Disorders In Children, Vitiligo And Albinism
Oct 12, 2023

Pigmentary disorders are the commonest group of dermatoses in paediatric age group. There are 2 types of pigmentary disorders: Hyperpigmentation and Hypopigmentation
Hyperpigmentation in Children
According to Nelson, this hyperpigmentation is usually reversible, and over some time, it tends to fade or disappear. Sun protection and controlling the un underlying inflammation can have a very good effect onpost-inflammatory hyperpigmentation.
COMMON CONDITIONS
- Café-au-lait-spots stands for coffee-coloured spots.
- Ephelides (freckles) are a type of isolated photosensitive lesions.
- Lentigines are seen in various syndromes including Lunan syndrome and Karmi complex.
1.CAFÉ-AU-LAIT SPOTS/CAFÉ-AU-LAIT MACULES ( CALMs )
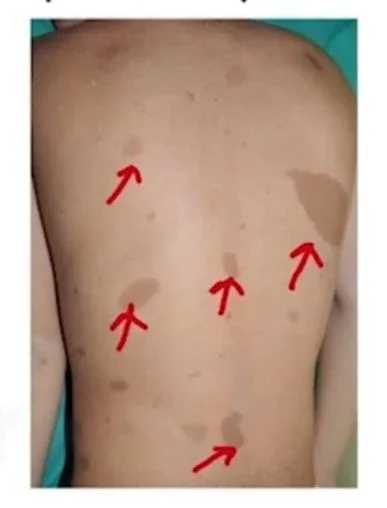
- They are brown-coloured/coffee-coloured macules that are well-defined, variable in size, and distributed all over the body. They can be on the photo-exposed part of the body or the non-photo-exposed part as well. 1 to 3 cafe-au-lait spots can be seen even in normal children. However, multiple spots are usually indicators of some underlying conditions. A concerning number of spots can be >6. In histopathology: There is an increase in the number of melanocytes and an increase in the melanin deposition in the epidermis. But, no enlargement of rete ridges can be seen.
| COMMON DISORDERS ASSOCIATED | |
| Neurofibrosis-1,2 | Strong association |
| McCune-Albright syndrome | Strong association |
| Cowden syndrome | Strong association |
| Ring chromosome syndrome | Strong association |
| Russell-silver syndrome | Weak associations |
| Basal cell nevus syndrome | Weak associations |
| Fanconi anaemia | Weak associations |
| Bloom syndrome | Weak associations |
| Chediak-Higashi syndrome | Weak associations |
| Tuberous sclerosis | Weak associations |
| Gaucher syndrome | Weak associations |
| Maffucci syndrome | Weak associations |
| Hunter syndrome, Turner syndrome | Weak associations |
2. Ephelides (Freckles)
- Ephelides are usually small (<3mm), brown hyperpigmented, macules that are present on the photo-exposed parts like face, upper back, dorsum of hands and feet. They are less sharply defined, and they tend to increase with photo exposure e.g., summer, and decrease e.g., winter. Histopathology: No increase in the overall melanin content in the skin. However, increase in melanin deposition can be seen in the basal epidermal calls. These basal epidermal cells tend to show an increase in branching as compared to normal children. Ephelides are more common in fair-skinned individuals as well as red hair one. Increase risk of developing malignant melanomas.

3.Lentigines
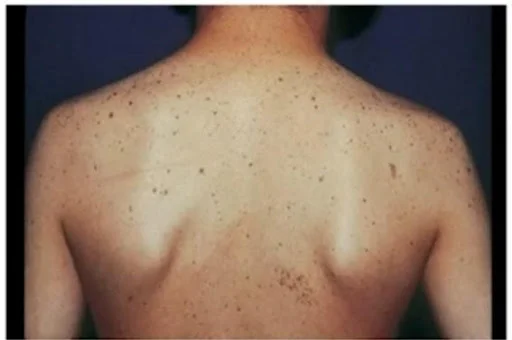
They are in the form of small (3-5 mm) brown macular lesions, they are sharply demarcated. They can be present anywhere on the body. They are not related to photo exposure. On histology, we can see that there is an increase in the number of melanocytes and club-shaped thickening of epidermal rete ridges.
Also Read: Aneuploidies Including Turner And Klinefelter Syndrome
Conditions Associated with Lentigines
- Lentiginosis profusa: there are multiple, extensive, lentigines with no mucosal involvement.
- LAMB syndrome (carney complex)- PORKAR1 gene mutation on 17q. Often have associated endocrine disorders like ACTH- independent Cushing syndrome. High risk of developing Sertoli cell cancers of the tests in males. LAMB stands for L: Lentigines of the face and vulva A: atrial myxoma M: mucocutaneous myxoma B: blue nevi.
- LEOPARD syndrome type1: PTPN-11 gene mutation and type2: RAF-2 gene mutation. LEOPARD syndrome is also called Noonan syndrome with multiple lentigines. LEOPARD stands for L: lentigines E: ECG abnormalities O: ocular hypertelorism P: pulmonary stenosis A: abnormal genitalia (hypogonadism, cryptorchidism, etc.) R: retardation of growth D: deafness (SNHL)
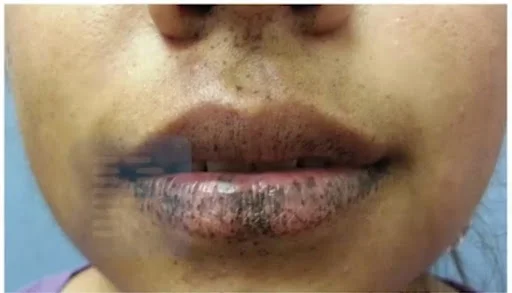
- Peutz-Jeghers syndrome: autosomal dominant- STK11 gene mutation. Hallmark of Peutz-Jeghers syndrome is lentigines on the oral mucosa.
Key Features of Lentigines
- They have an increased risk of lentigines, frequently present in the oral cavity, lips, perioral, hands and feet. They have a high risk of developing polyps, which tend to occur most commonly in the small intestine> stomach> colon, etc. In the small intestine the most common site is jejunum followed by ilium. These polyps can produce features like abdominal pain, and bleeding. Intercuspation can develop due to an underlying polyp. Increased risk of malignancy can be both: GIT malignancy as well as non-GIT malignancy. GIT malignancy can be in the form of colorectal cancer and can also occur in the form of other cancers namely small intestinal cancers, stomach cancers, and anal cancers. In non-GIT, testicular cancers are common, and ovarian cancers can also be seen. Sometimes, epididymis benign and malignant tumours can also be seen, but they are rarer.
Also Read: KAWASAKI DISEASE : History, Symptoms, Causes and Treatment
Hypopigmentation in Children
Common Condition
- Piebaldism
- Waardenburg syndrome
- Tuberous sclerosis
- Hypomelanosis of ITO
- Albinism
- Vitiligo
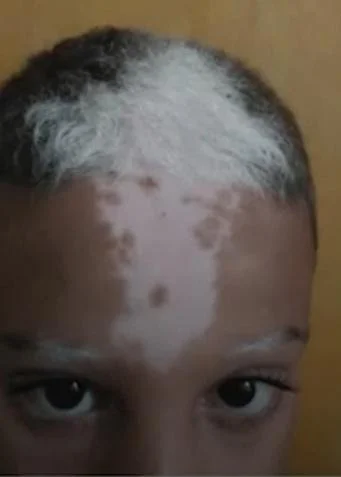
- Piebaldism
- It is an autosomal dominant condition that shows c-KIT receptor defect. Characterized by sharply demarcated amelanotic patches. Common sites are the forehead, and anterior scalp- producing a white forelock, ventral trunk, knees and elbows. Islands of normal or darker-than-normal pigmentation may be present within the amelanotic areas.
2. Waardenburg Syndrome
- A condition characterized by localized areas of depigmented skin and hair. Shows autosomal dominant inheritance. There are mainly four types of Waardenburg syndrome.
- Type1: heterogenous PAX3 gene mutations
- Hallmark is white forelock. Depigmented skin in only 15%. Dystopia Canthorum i.e., Telecanthus in all patients. Unibrow i.e., Synophrys in 17-69%. Deafness in 9-37%. Heterochromia irides in 20%.
- What is white forelock?
- White Forelock is part of a group of conditions which is known as poliosis, which is the absence of melanin in a specific segment of skin or hair. When poliosis affects a tuft of hair just above the forehead i.e., called white forelock.

Type2: MITF gene mutations
- Similar to type I, except no Telecanthus and deafness more common.
Type3: homozygous PAX3 gene mutations.
- Also called Klein Waardenburg syndrome. Type1 + limb abnormalities, e.g., contractures, and hypoplasia.
Type4: multiple genes- SOX10, EDNRB and EDN3.
- Also called Shah Waardenburg syndrome. Type1+ Hirschsprung disease. Telecanthus is only seldom seen.
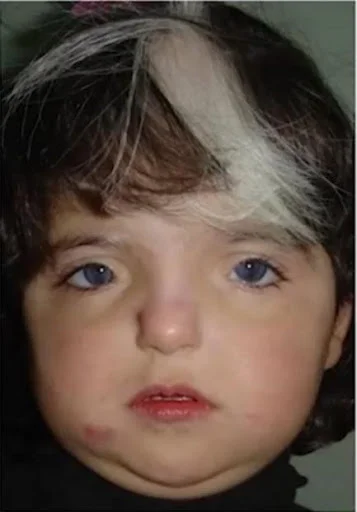
3. Tuberous Sclerosis
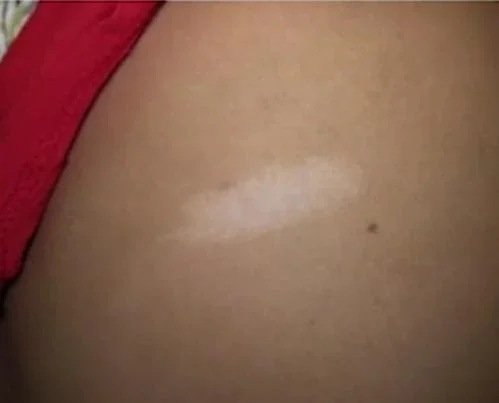
One of the skin manifestations of tuberous sclerosis is ash-leaf macules. They are commonly present on the trunk, they are hypopigmented, irregular to regular in shape and they are best seen by wood’s UV lamp. Hypopigmented lesions on the trunk or limbs along with seizures especially, hypsarrhythmia on EEG the case will be tuberous sclerosis.
4. Hypomelanosis Of Ito
Also known as Blaschkoid or mosaic hypomelanosis. Lesions are present since birth or appears within 2 years of life. Morphology is in the form of bizarre, patterned, hypopigmented macules arranged in sharply demarcated whorls/ streaks/ patches. Lesions follow the lines of Blaschko. They tend to spare palms, souls, and oral mucosa. In Histology: There is fewer and smaller melanocytes and a decreased number of melanin granules in the basal cell layer than normal. No inflammatory cells.
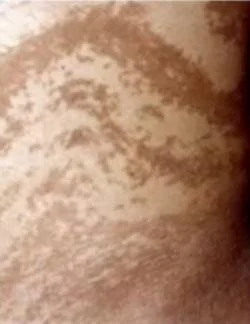
Associated Abnormalities
The most common are in the central nervous system. There are intellectual dysfunctions, seizures, and microcephaly or hypotonia. Other abnormalities are musculoskeletal system. There can be scoliosis > limb deformities. Other abnormalities include eye abnormalities like strabismus, and nystagmus in some patients. There is an increased risk of cardiovascular disorder e.g., congenital heart disease.
Difference Between Vitiligo And Albinism
5. Albinism
- Albinism refers to a relative or absolute deficiency of melanin in the skin or eyes or other pigmented organs.
Pathophysiological Basis Of Albinism
- Pigment in the body which gives a dark color to the body is known as melanin.Melanin is of broadly two types:
- Pheomelanin
- Eumelanin.
- Pheomelanin is reddish-yellow in color.
- Eumelanin is brown-black in color.
- Melanin is formed from an amino acid called tyrosine. There are several enzymes involved but the most prominent one is known as the tyrosinase enzyme. There are neural crest cells, and there are a set of cells called melanoblasts. These melanoblasts migrate to various organ structure and produces melanocytes cell. These cells are predominantly present in the dermo-epidermal junction of the skin. They are present in the hair follicle, internal ear (cochlea), and ocular structures (RPE and in the iris).

Definition And Traditional Classification
- Albinism is characterized by a deficiency of melanin in various pigmented structures. Broadly, there are two types of albinism; Generalized albinism, and Localized albinism. Generalised can be divided further into two parts: oculocutaneous albinism and ocular albinism.
Oculocutaneous albinism is autosomal recessive in nature. Ocular albinism is an X-linked recessive inheritance. Localized forms consist of piebaldism and Waardenburg syndrome. In the generalized form there is a syndromic form of generalized albinism. Hermansky-Rudlak syndrome and Chediak-Higashi syndrome are examples of this.
Clinical Features Of Albinism
- General appearance: Pale white skin, blond-white hair, blue coloured eye. Ocular manifestations: These children will have an increased risk of photophobia. Also, there will be hypopigmentation of the iris. Hypopigmentation of the retina with foveal hypoplasia. Reduced visual acuity. Refractive errors can be seen. Nystagmus can also be present. Iris transilluminations i.e., reddish hue on ophthalmoscopic/slit lamp examination.
Visual Field Defects In Albinism
- In albinism, most temporal side fibres cross to opposite sides in albino patients so they have a lack of perception and alternating strabismus.
Individual Subtypes Of Albinism
- Oculocutaneous Albinism
- OCA type1/OCA1: aka tyrosinase-deficient albinism.
- TYR gene mutation on 11q.
- Subtypes:
- OCA1A- Most severe form, no tanning, no nevi, no freckles
- OCA1B is considered to be a relatively milder form.
- OCATS - cooler regions of the body may show some pigmentation.
- OCA type2/OCA2 tyrosinase-positive albinism.
- Overall, most common type of generalized OCA- some pigment, some tanning. OCA2 gene mutation on 15q. May also be associated with Prader Willi and Angelman syndrome.
- OCA type3/OCA3: Rufous albinism.
- TYRP1 gene mutation on 9p. Can make pheomelanin but not eumelanin. Red hair and red crown skin are present.
- OCA Type 4/OCA4.
- Similar to OCA2, more in Japan. SLC45A2 (previously called MATP) gene mutation on 5p.
- OCULAR ALBINISM
- X-linked recessive- GPR143 gene on Xp. Most common variety- OA1 also called Nettleship-Falls OA. Males with prominent ocular features but normal to slightly less skin pigmentation. EM: macro melanosomes in skin biopsies or hair root specimens.
Syndromic Forms Of Generalized Albinism
- Characterized by defects in melanosomes.
- Hermansky-Pudlak syndrome
- Is characterized by autosomal recessive inheritance. There are 9 types of HPS genes. These patients will have OCA (similar to OCA2 variety) + bleeding tendency due to platelet defects. They will have a high risk of developing pulmonary fibrosis, renal failure, cardiomyopathy etc.
- Chediak-higashi syndrome.
- There is a mutation in the LYST gene. They are intracellular trafficking genes. They will have an increased risk of infections + bleeding manifestation will be seen + oculocutaneous albinism can be seen.
- Griscelli syndrome is a combination of albinism with intellectual dysfunction.
- Key points to know are: Sun protection is a must with sunscreen with SPF >30. And they need to be monitored by ophthalmoscopic monitoring and screening for UV-associated malignancies, basal cell carcinoma, and squamous cell carcinoma seen in these patients.
6. VITILIGO
- Vitiligo is an acquired macular depigmentation disorder associated with the destruction of melanocytes. Affects 0.5-2% of the population worldwide. Males and females are equally affected. Positive family history is found in 20-30% of cases.
Pathophysiologic basis:
- There is almost 80% of autoimmune melanocyte destructions. Most of the patients are found to have anti-melanocytes antibodies. They are also found to have CD8+ t-cells directed against melanocytes.

Types of vitiligo
- Non segmental (80-90%)- Koebner phenomenon
- Segmental (10-20%)- no Koebner phenomenon
Management Of Vitiligo
- In case it is a non-segmental form and if it is a localized thing, topical steroid can be used or topical tacrolimus also can be used in the segmental form. In generalized non-segmental form, oral steroids are given in a short course + phototherapy in the form of narrow-band UVB (NB-UVB). It is also called the therapy of choice in widespread lesions, also written as (UVB-311). PUVA therapy can also be used (Psuralen + UVA).
Also Read: 5 pro tips to unleash peak performance in the NEET-SS exam.
Hope you found this blog helpful for your NEET SS General Pediatrics preparation. For more informative and interesting posts like these, keep reading PrepLadder’s blogs.

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Navigate Quickly
Hyperpigmentation in Children
1.CAFÉ-AU-LAIT SPOTS/CAFÉ-AU-LAIT MACULES ( CALMs )
Conditions Associated with Lentigines
Key Features of Lentigines
Hypopigmentation in Children
Common Condition
Associated Abnormalities
Difference Between Vitiligo And Albinism
Pathophysiological Basis Of Albinism
Definition And Traditional Classification
Clinical Features Of Albinism
Visual Field Defects In Albinism
Individual Subtypes Of Albinism
Syndromic Forms Of Generalized Albinism
Types of vitiligo
Top searching words
The most popular search terms used by aspirants
- NEET SS Pediatrics