Sickle Cell Anemia And Thalassemia
Apr 11, 2023

Sickle cell disease and thalassemia are genetic disorders and they are caused by errors in the genes for hemoglobin. These are essential topics under Pathology so feel free to revisit this blog post as many times as necessary.
Read the following post thoroughly and level up your NEET PG Pathology preparation.
- Hemoglobinopathies - Sickle cell anemia and Thalassemia.
- Normal hemoglobin
Adult hemoglobin (HbA) is 95% and above and it is made of α 2 and β 2 chains. HbA2 is Less than 3% and is made of α 2 and delta 2 chain whereas Fetal hemoglobin (HbF) is made of α 2 and gamma 2 chain.

1. Sickle Cell Anemia
- Mutation - Point mutation
Int this the Problem occurs at β 6 subunit. Glutamic acid is changed to valine. When glutamic acid is present - patients have HbA. When changed to valine the HbA changes to sickled hemoglobin (HbS). So, it is a missense point mutation.
| At β 6 position, if glutamic acid changes to lysine. It is called HbC hemoglobin disorder. 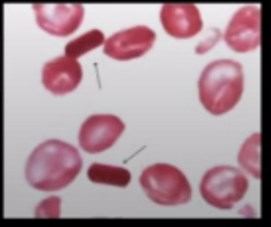 |
Factors of Sickle Cell Anemia
Amount of HbS and HbA- HbS is directly proportional to Sickling, it means more are the levels of hbs more sickling of the cells will be present.
- Cell converts to Sickle cell when:
- Normal O2 levels are low - Hypoxia.
- Less PH - Acidic environment.
- Less water - Dehydration.
- Dehydration: More sickling.
- Hypoxia: More sickling.
- Less PH: More sickling.
- Amount of fetal hemoglobin- Fetal hemoglobin has more affinity to oxygen. So when more oxygen levels are present we will find less sicking.
- Sickle-alpha thalassemia- In alpha thalassemia there is no α chain so there is formation of Less hemoglobin. So as there is less hemoglobin so we will find less sickling of the cells.
Types
- HbAA - Normal.
- HbAS - Heterozygous type.
- Sickle cell trait.
- HbSS - Homozygous type.
- Sickle cell anemia.
- Sickle cell anemia is autosomal resistance disease.
Compound Heterozygous
It Contains both harmful genes Like HbβS, Here both the genes are harmful. β thal and Sickle cell genes are present. If it contains one normal and one harmful gene then this condition is known as heterozygous.
Pathophysiology
Regular Normal cells
In the absence of oxygen and less pH The cells convert to sickle shape . This is a reversible process. When there is enough oxygen and proper PH, the sickle shaped cells are converted to normal again and this condition is known as called reversible sickling. But When there is repeated reversible sickling it leads to irreversible sickling. These sickle shaped cells are very sticky. They stick to each other and forms a mass. If these get stuck in the narrow blood vessels - results in vaso-occlusive crisis.
Clinical Features of Sickle Cell Anemia
- Pallor
- Splenomegaly - It is an extravascular hemolysis and the cells are broken by spleen, Causing splenomegaly.
- Jaundice - RBCs are broken therefore increases the level of bilirubin.
- Vaso-occlusive crisis in different organs
- Brain - results in stroke.
- Heart - results in MI.
- Lungs - results in acute chest syndrome.
- Bones
- Vertebra - Fish mouth vertebra.
- Femur - Avascular necrosis.
- Digits - Acute dactylitis.
|
Other Crisis of Sickle Cell Anemia
- Aplastic crisis: Associated with parvovirus B19 infection.
- Hemolytic crisis: Associated with Epstein Bar Virus. In this condition Everything is broken off.
- Sequestration crisis: All sickle cells are sequestered into the spleenand then these cells Attacks the spleen and damages it. This is Referred as multiple splenic infarcts and it leads to decrease in spleen size which is known as Auto splenectomy.
Initially the spleen gets enlarged as it is performing more functions. When a sequestration crisis forms - the spleen gets shrinked. Leads to auto splenectomy. |
Also Read:
CHRONIC INFLAMMATION: Symptoms, Causes, Diagnosis, and Treatment: Pathology
Diagnosis of Sickle Cell Anemia
- Hemoglobin: Hb levels decrease
- Reticulated Count: As bone marrow works more the reticulocyte count increases.
- Bilirubin: As it is an extravascular hemolysis the breakdown of RBCs increases bilirubin.
- ESR: Erythrocytes Sedimentation Rate. Normal cells get packed and settle down whereas the Sickle cells do not get packed and are not settled down. Hence, ESR is low.
Sickle-shaped cells
These are also called drepanocytes. It is Only seen in sickle cell anemia. Sickle-shaped cells are seen only in sickle cell anemia but not in sickle cell trait.

a. Sickling Test
It is Done if there are no sickle cells to know if it is sickle cell trait or not.
Sickling test
Take a clean slide and Add a drop of patients blood. Add 2% sodium metabisulfite or sodium dithionite To remove oxygen from the cells. And then Cover the slide with coverslip to prevent the exposure of atmospheric oxygen then seal the coverslip with paraffin wax or nail paint. The cells convert to sickle shape. This test is very primitive. This test Do NOT differentiate between sickle cell anemia and sickle cell trait.
b. Solubility Test
Sickle cells are sticky and also have less solubility. It is also a primitive test. In this Blood is added into the test tube and then sodium dithionite is added to remove oxygen. Sickle cells form polymers, which makes the test tube turbid which indicates less solubility.
c. Hemoglobin Electrophoresis
- It is of 2 types
- Cellulose acetate electrophoresis - pH 8.4 (alkaline).
- Citrate electrophoresis - pH 6.2 (acidic).
- Cellulose acetate electrophoresis
- Hemoglobin electrophoresis has 2 ends - anode and cathode.
- Hemoglobin bands are formed.
- Hemoglobin H
- Hemoglobin A
- Hemoglobin F
- Hemoglobin S
- Hemoglobin A2
- Mnemonic: HAFSA2
- These bands are formed from anode to cathode. Along with hemoglobin S, hemoglobin D can also be seen. To differentiate these the other type citrate electrophoresis is used.
- Citrate electrophoresis
- Here the hemoglobin S and D are splitted which are formed in cellulose acetate electrophoresis along With Hemoglobin A2 - C, E, O-Arab.
Note: Hemoglobin A2, C, E, O-Arab are present near the cathode.
Concept - Slide is taken and blood is placed at cathode
- If hemoglobin moves faster and reaches the anode - HbA (normal).
- If hemoglobin presents both towards anode and cathode - HbAS (sickle cell trait).
- If hemoglobin moves slower and stays at cathode - HbS (sickle cell anemia).
- Even the electrophoresis doesn't show proper results as there are overlaps and no quantity is examined.
d. HPLC: High-Pressure Liquid Chromatography.
- Gold standard test for any type of hemoglobin disorders.
HPLC graph
- Graph shows different peaks for different hemoglobin.
| Peak Name | Retention time, Min |
| P1 window | 0.63-0.85 |
| F window | 0.98-1.20 |
| P2 window | 1.24-1.40 |
| P3 window | 1.40-1.90 |
| A0 window | 1.90-3.10 |
| A2 window | 3.30-3.90 |
| D window | 3.90-4.30 |
| S window | 4.30-4.70 |
| C window | 4.90-5.30 |
- Peak name and retention time
- The peaks are formed based on the elution time.
Treatment of Sickle Cell Anemia
- As it is a hereditary disorder there is nothing more can be done.
- Hydroxyurea (to treat hypoxia)
- Increases the fetal hemoglobin.
- Therefore increases oxygen affinity.
2. Thalassemia
Adult hemoglobin is Made up of α2 and β2 chains.This means there are 2 alpha chains and 2 beta chains. 4 alpha genes are present on chromosome 16 and 2 beta genes present on chromosome 11 which gives 2 alpha chains and 2 beta chains respectively . Absence of α gene leads to alpha thalassemia and absence of β gene leads to β thalassemia.
Note
- α thalassemia - There is α gene deletion.
- β thalassemia - There is β gene mutation.
Pathology Related Articles:
A. β Thalassemia
- 3 types
- Thalassemia major - It is always dependent on transfusion. It is also known as Cooley anemia.
- Thalassemia intermedia
- Thalassemia minor / trait - Heterozygous
|
Feature |
Thalassemia major |
Thalassemia intermedia |
Thalassemia minor / trait |
|
β chain production |
|
|
|
|
Clinically |
|
|
Asymptomatic |
|
Hb |
3 to 5 (more severe) |
5 to 8 |
> 8 |
|
Iron profile |
Increased |
Normal |
Normal |
|
Hb electrophoresis and HPLC |
Increased HbF |
Both increased |
HbA2 > 3.5% |
- β thalassemia is also an extravascular hemolytic disease.
β Thalassemia - Genetics
- Most of them are promoter region mutation.
- Splicing mutation can also occur - most common.
- Splicing - removes introns.
- Mutation in intervening sequence - IVS1-5 G to C in India.
- Chain termination mutation.
- Frameshift mutation: Occurs at +8/9th codon or +41/42 codon.
| Only deletion in β thalassemia - 619 base pair deletion. |
a. Pathogenesis of β Thalassemia Major
In this condition the β chains are missing. There are no β chains available for α 2 chains to combine. The α 2 chains to sustain and maintain the valence of 4 - tires 2 possibilities:
It stays alone and forms α 4 tetramers. This leads to ineffective erythropoiesis which Increases load on bone marrow. This is known as Erythroid hyperplasia.
It combines with gamma 2 (fetal hemoglobin). This has high affinity for oxygen. Less oxygen is reached to tissues . Stimulates the release of erythropoietin. Increases work load on bone marrow - Erythroid hyperplasia
- Therefore, in thalassemia major:
- HbF increases
- RBC increases but Hb decreases - Typical in thalassemia patients.
- Iron levels increase.
As there is erythroid hyperplasia so it requires some energy to work more. It Stimulates the GIT to absorb more Iron so the Iron levels increases. The patients are kept on blood transfusions. Repeated blood transfusion also increases Iron levels and it leads to Iron overload. Erythroid hyperplasia increases work on other bones and organs as well . It stimulates erythropoiesis from other bones and organs. This erythropoiesis from other bones leads to Crew Cut Skull. The erythropoiesis from other organs is known as Extra medullary Hematopoiesis.
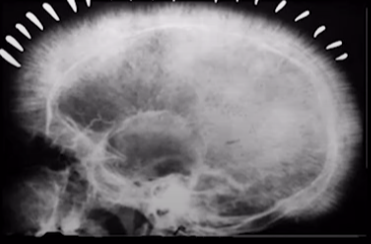
Crew cut appearance of skull
- This is also called Hair on End appearance. It is seen in both thalassemia and sickle cell anemia. This is an indication of erythropoiesis in other bones.
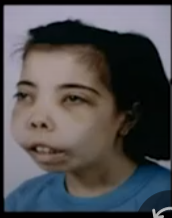
Chipmunk facies
- Maxillary Prominence is present.
Peripheral Smear Finding of β Thalassemia Major
- It is a type of microcytic hypochromic anemia. In this Nucleated RBCs are present. Target cell or codocyte is also present.
- Confirmatory test - HPLC.
- Shows high HbF.
Note: Globin gene sequencing is the best technique.
b. β Thalassemia Minor/Trait
- Microcytic hypochromic anemia.
- Asymptomatic.
- The condition is confused with iron deficiency anemia.
| Feature | Iron Deficiency Anemia | β Thalassemia Minor |
| Type | Microcytic hypochromic anemia | Microcytic hypochromic anemia |
| RBC | Less | More |
| RDW (size variance) | More | Normal |
| Mentzer index: MCV/RBC | > 13 | < 13 |
| HbA2 - confirmatory test | <3.5% | > 3.5% |
| \Iron deficiency anemia is associated with dietary changes. Less Iron in diet - Size of RBC decreases. More Iron or sufficient Iron - Size becomes normal. Thalassemia Is associated with defects in genes. |
Also Read:
Henoch- Schonlein Purpura : Causes, Symptoms, Pathophysiology, Diagnosis, Treatment and Prognosis
Trisomy 18: Causes, Symptoms, Types, Diagnosis, Treatment and Prevention : Pathology
Nestroft Test
This test is used as One of the screening tests for β thalassemia. It is the Naked Eye Single Tube RBC Osmotic Fragility test. Thalassemia cells are always tough. No microscope is needed to identify the results. The test is used to test osmotic fragility of RBCs.
Method of Nestroft Test
In this 2 test tubes are taken and then the patient's blood and normal person's blood is added in each 5 ml of 0.35% normal saline to both making a Hypotonic solution. The hypotonic saline starts entering the RBCs. A white paper with black line is placed behind both the tubes. The RBCs of normal person blood breaks due to hypotonic saline and the black line is visible whereas Thalassemia cells do not break and the line is not visible.

- Mnemonic: ThALESSemia
- A - absent lines
- LESS - Mentzer index is < 13.
B. α Thalassemia
In this condition the alpha chains are missing due to α gene deletion.
|
α Gene Detection |
State |
|
AA/AA - All 4 are present |
Normal |
|
AA/A- 1 α gene is deleted |
Asymptomatic |
|
AA/- - 2 α genes are deleted |
α thalassemia trait |
|
A-/- - 3 α genes are deleted |
|
|
All 4 α genes are deleted (- -/- -) |
|

Golf ball inclusions
- New methylene blue stain is used. The blue ball-like cells indicate golf ball inclusions. These are due to β 4 tetramers.It is Seen in 3 alpha gene deletion thalassemia which is known as HbH disease.
- Mnemonic: He is a Boy playing Golf at 3 AM
- He - HbH
- Boy - β 4 tetramers
- Golf - Golf ball inclusions
- 3 AM - 3 α gene deletion
Also Read : Amyloidosis: Types, Causes, Symptoms, Diagnosis, Risk Factors, Treatment
And that is it! You have now covered everything you need to know about Sickle cell disease and Thalassemia for your Pathology paper preparation. For more interesting and informative posts like these, download the PrepLadder App keep following our blog.

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Navigate Quickly
1. Sickle Cell Anemia
Factors of Sickle Cell Anemia
Types
Compound Heterozygous
Pathophysiology
Clinical Features of Sickle Cell Anemia
Other Crisis of Sickle Cell Anemia
Diagnosis of Sickle Cell Anemia
Sickle-shaped cells
a. Sickling Test
Sickling test
b. Solubility Test
c. Hemoglobin Electrophoresis
d. HPLC: High-Pressure Liquid Chromatography.
HPLC graph
Treatment of Sickle Cell Anemia
2. Thalassemia
Note
A. β Thalassemia
β Thalassemia - Genetics
a. Pathogenesis of β Thalassemia Major
Peripheral Smear Finding of β Thalassemia Major
b. β Thalassemia Minor/Trait
Nestroft Test
Method of Nestroft Test
B. α Thalassemia
Golf ball inclusions
Top searching words
The most popular search terms used by aspirants
- NEET PG Pathology
- NEET PG Preparation